
유한양행의 폐암 신약 ‘렉라자’(성분명 레이저티닙)와 미국 얀센의 ‘리브리반트’(성분명 아미반타맙) 병용요법에 대한 미국 식품의약국(FDA) 허가 결과가 8월 중 나올 것으로 전망된다. 최근 열린 미국임상종양학회(ASCO)에서 긍정적인 임상결과를 공개해 허가 가능성이 높아졌다는 분석이다.
16일 업계에 따르면 렉라자와 리브리반트 병용요법 FDA 허가 여부는 8월 21일 이전에 발표될 예정이다. 앞서 유한양행은 2018년 존슨앤드존슨(J&J)에 렉라자를 12억5500만 달러(약 1조4000억 원) 규모로 기술이전했다. 존슨앤드존슨 자회사 얀센은 지난해 말 FDA에 비소세포폐암(NSCLC) 1차 치료제로 해당 병용요법에 대한 신약허가신청서(NDA)를 제출했다.
FDA 승인 문턱을 넘으면 해외 판매 로열티와 단계별 마일스톤(기술료)까지 확보할 수 있어 유한양행에 호재로 작용할 전망이다. 승인 완료 시 유한양행은 올해 4분기 중 얀센으로부터 6000만 달러(약 826억 원)의 마일스톤을 받게 된다.
임상결과는 긍정적이다. 유한양행과 존슨앤드존슨은 ASCO 2024에서 렉라자와 리브리반트의 병용요법인 ‘마리포사(MARIPOSA)’의 임상 3상 후속 연구결과를 공개했다. 전이와 변이 가능성이 큰 고위험 환자군에 병용 투여했을 때 종양 진행 정도와 사망 위험을 낮춘다는 결과를 제시했다.
리브리반트를 정맥주사(IV)에서 피하주사(SC) 제형으로 변경한 임상인 팔로마-3(PALOMA-3)도 주목을 받았다. 제형 변경으로 투약시간이 기존 4~5시간에서 5분으로 단축했고, 주입 관련 반응(IRR) 부작용이 66%에서 13%로 현저히 감소했다. 정맥혈전색전증(VTE) 부작용도 일부 개선되는 효과를 확인했다.
팔로마-3 임상은 올해 최고 발표(Best of ASCO)로 선정됐다. 얀센은 해당 임상결과를 바탕으로 유럽의약품청(EMA)에 ‘렉라자+리브리반트’ SC제형 승인 신청서 제출을 완료했다. 향후 미국을 포함한 다른 국가에도 승인 신청서를 제출할 것으로 보인다.
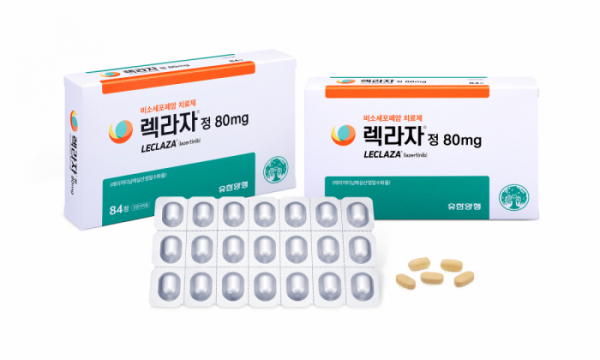
렉라자가 FDA 허가를 받으면 국내 제약사가 개발한 후보물질이 글로벌 빅 파마에 기술이전해 제품 출시까지 하는 첫 번째 사례가 된다. 렉라자는 이미 한국에서, 리브리반트는 미국에서 각각 단독요법 1차 치료제로 승인받았기 때문에 업계는 두 약물 병용요법의 FDA 허가 가능성은 높다고 전망한다.
긍정적인 임상결과가 꾸준히 나오고 있는 만큼 유한양행의 실적 전망과 기업 가치가 오르고 있다. 박재경 하나증권 연구원은 최근 보고서에서 “렉라자의 파이프라인 가치를 기존 1조9500억 원에서 2조5000억 원으로 약 25% 상향 조정한다”고 했다. 또 유한양행의 올해 실적으로 연매출이 2조 원을 넘길 것이란 증권가 전망도 나왔다.
하현수 유안타증권 연구원은 “렉라자+리브리반트 병용요법이 타그리소(성분명 오시머티닙) 단독 요법에 비해 전이와 추가적인 변이가 있는 고위험 환자군에서 위험 감소가 더 크게 나타나 중심이 될 것”이라며 “비소세포폐암 환자는 진단 당시 다수의 환자가 전이성을 보이는 등 고위험환자군의 비중이 높은 암종이다. 이번 임상을 통해 렉라자+리브리반트 병용요법의 경쟁력을 강화했다”고 평가했다.
한편, 한국과학기술정보연구원(KISTI)에 따르면 2022년 비소세포폐암 치료제 글로벌 시장 규모는 279억5200만 달러(37조 원)에서 매년 연평균 13%씩 성장해 2026년 427억1200만 달러(56조 원) 규모로 커질 것으로 전망됐다.
![[단독]내일부터 암, 2대 주요치료비 보험 판매 중지된다](https://img.etoday.co.kr/crop/140/88/2105043.jpg)
!["아이 계정 삭제됐어요"…인스타그램의 강력 규제, '진짜 목표'는 따로 있다? [이슈크래커]](https://img.etoday.co.kr/crop/140/88/2105286.jpg)
![근무시간에 유튜브 보고 은행가고…직장인 10명 중 6명 '조용한 휴가' 경험 [데이터클립]](https://img.etoday.co.kr/crop/140/88/2105320.jpg)

![[단독] LG 생성형 AI ‘엑사원’에 리벨리온 칩 ‘아톰’ 적용되나…최적화 협업 진행](https://img.etoday.co.kr/crop/140/88/2105118.jpg)
![[인터뷰] 조시 팬턴 슈로더 매니저 “K-채권개미, 장기 투자로 美은행·통신·에너지 채권 주목”](https://img.etoday.co.kr/crop/140/88/2105216.jpg)







![“만두 1위 굳히기”…CJ제일제당, 미국·헝가리에 비비고 공장 세운다 [종합]](https://img.etoday.co.kr/crop/85/60/2105188.jpg)
![‘K뷰티 랜드마크’ 성수 상륙...올리브영N엔 건강·美 가득[가보니]](https://img.etoday.co.kr/crop/85/60/2105193.jpg)





![[정치대학] 이재명 대안은 김부겸·김동연?…박성민 "둘 다 명분 없다"](https://img.etoday.co.kr/crop/300/170/2105457.jpg)
![서울 명동 임대료, 세계 9번째로 '비싸' [포토]](https://img.etoday.co.kr/crop/300/190/2105411.jpg)